When you pick up a generic pill at the pharmacy, you expect it to work just like the brand-name version. But how do we know it really does? The answer lies in pharmacokinetic studies - the most widely used method to prove that a generic drug behaves the same way in your body as the original. Yet, calling it the "gold standard" is misleading. It’s not perfect. It’s not always enough. And for some drugs, it’s downright inadequate.
What Pharmacokinetic Studies Actually Measure
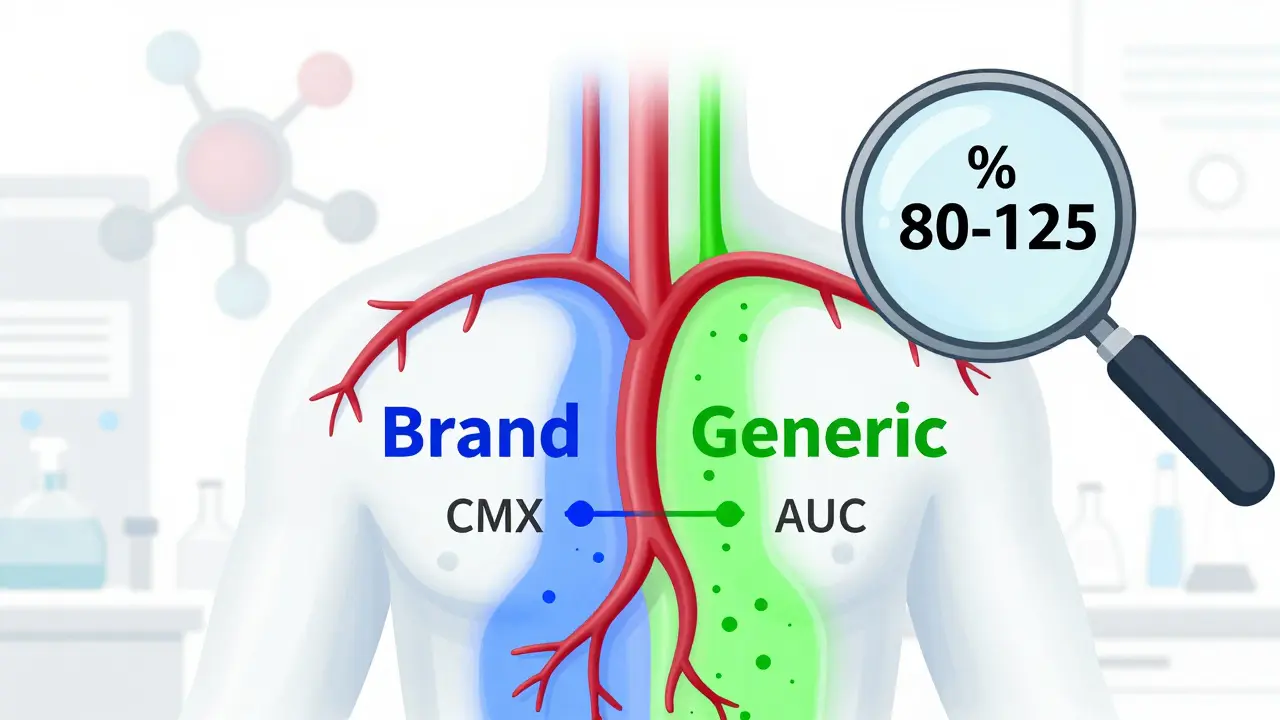
Pharmacokinetic studies track how a drug moves through your body. Specifically, they measure two key numbers: Cmax (the highest concentration of the drug in your blood) and AUC (the total amount of drug your body absorbs over time). These aren’t just lab curiosities. They tell us whether the generic drug gets into your bloodstream at the same rate and to the same extent as the brand-name version.
The U.S. Food and Drug Administration (FDA) requires that the 90% confidence interval for the ratio of generic to brand-name drug falls between 80% and 125% for both Cmax and AUC. That means if the brand drug delivers 100 units of drug into your system, the generic must deliver between 80 and 125 units. Anything outside that range? It’s not approved.
These tests are done in healthy volunteers - usually 24 to 36 people - using a crossover design. Each person takes both the generic and the brand-name version at different times, with a washout period in between. This removes individual differences in metabolism from the equation. Studies are often done both fasting and after eating, because food can change how a drug is absorbed. For example, a statin like simvastatin might absorb poorly on an empty stomach but much better with a meal.
Why This Isn’t the "Gold Standard" - Even Though Everyone Says It Is
The FDA itself says bioequivalence isn’t a gold standard. It’s a scientifically validated surrogate. That’s a fancy way of saying: we use these blood tests because they’re practical, repeatable, and correlate well with clinical outcomes - but they don’t measure the actual effect on your disease.
Take warfarin, a blood thinner with a narrow therapeutic index. A tiny difference in absorption can mean the difference between a stroke and a bleed. For these drugs, the FDA tightens the acceptable range to 90-111%. Even then, there have been cases where generics passed pharmacokinetic tests but still caused problems in real patients. Why? Because pharmacokinetics doesn’t capture everything. It doesn’t tell you how the drug interacts with other medications, how your liver processes it over time, or whether inactive ingredients (excipients) trigger an immune response.
And here’s the kicker: two generics made by different companies, both approved using the same pharmacokinetic data, can behave differently in practice. A 2010 PLOS ONE study found that two generics of gentamicin - both identical in chemical composition and in vitro tests - had wildly different pharmacodynamic profiles in patients. One worked. The other didn’t. The pharmacokinetic study didn’t catch it.
Where Pharmacokinetic Studies Fail: Topical, Complex, and NTI Drugs
For creams, ointments, inhalers, or injectables, measuring blood levels tells you almost nothing. A steroid cream might be absorbed through your skin at different rates depending on the base formula - even if the active ingredient is identical. You can’t measure that with a blood draw.
That’s why dermatopharmacokinetic (DMD) methods are gaining traction. These use microdialysis or tape-stripping to measure how much drug actually penetrates the skin. One 2014 study showed that in vitro permeation testing using cryopreserved human skin was more accurate and less variable than trying to run a clinical trial on hundreds of patients with eczema.
Same goes for modified-release tablets. A pill designed to release drug slowly over 12 hours can fail bioequivalence testing if the excipients change slightly. A different polymer coating might make the drug release too fast in the stomach, too slow in the intestine - or worse, unevenly. Pharmacokinetic studies might still show an AUC within range, but the peak concentration could spike dangerously early. That’s why the FDA now has over 1,800 product-specific guidances - each tailored to a unique drug’s behavior.

The Hidden Cost and Time of Getting a Generic Approved
Running a bioequivalence study isn’t cheap. It costs between $300,000 and $1 million. It takes 12 to 18 months. And that’s just the clinical part. Before you even get to human testing, you need to develop a formulation that matches the original. That often takes years of trial and error.
Manufacturers face a brutal trade-off: spend millions to prove equivalence, or risk rejection. Many companies abandon projects when they hit a wall with bioavailability. That’s why some generic drugs simply don’t exist - not because no one wants to make them, but because the science won’t cooperate.
There’s a workaround: the Biopharmaceutics Classification System (BCS). If a drug is highly soluble and highly permeable (BCS Class I), and the formulation is very similar, the FDA may waive the human study entirely. But only about 15% of drugs qualify. For the rest? You’re stuck with the blood tests.
Global Differences: FDA vs. EMA vs. WHO
The rules aren’t the same everywhere. The FDA takes a flexible, product-by-product approach. If your drug is complex, they’ll give you specific guidance. The European Medicines Agency (EMA), on the other hand, often uses a one-size-fits-all model. That creates headaches for global manufacturers trying to meet both standards.
The WHO recommends a broader definition of therapeutic equivalence: "effects, with respect to both efficacy and safety, are essentially the same." That means they accept clinical trials, pharmacodynamic studies, or even in vitro tests - depending on the drug. But many low- and middle-income countries lack the labs or expertise to run anything beyond basic pharmacokinetic studies. So they rely on what’s easiest - even if it’s not the most accurate.
ICH M13A, a harmonized guideline adopted by 35 countries, tries to unify methods for immediate-release oral drugs. But even that doesn’t cover complex products. The result? A patchwork of standards. A generic approved in the U.S. might not be accepted in Brazil. One approved in India might not get past the EMA.

The Future: PBPK Modeling and In Vitro Alternatives
The field is changing. Physiologically-based pharmacokinetic (PBPK) modeling uses computer simulations to predict how a drug will behave in the body - based on its chemical properties, organ function, and absorption pathways. Since 2020, the FDA has accepted PBPK models to waive bioequivalence studies for certain BCS Class I drugs. It’s not perfect, but for simple, well-understood drugs, it’s faster, cheaper, and sometimes more reliable.
And then there’s the growing evidence that in vitro tests - like dissolution profiles or permeability assays - can be more predictive than human studies. A 2009 PMC paper argued that for some immediate-release drugs, a well-designed lab test was more consistent than a small human trial with high variability. Why? Because human biology is messy. Lab conditions aren’t.
For topical drugs, DMD and IVPT are becoming standard. For inhalers, aerodynamic particle size analysis is replacing blood tests. The trend is clear: we’re moving away from a one-size-fits-all model. We’re moving toward smarter, targeted methods based on the drug’s chemistry, route, and risk.
What This Means for You
If you’re taking a generic drug for high blood pressure, diabetes, or depression - chances are, the pharmacokinetic study did its job. These drugs have been tested thousands of times. The system works.
But if you’re on warfarin, levothyroxine, or phenytoin - or using a topical steroid, an inhaler, or a long-acting injectable - you should be more cautious. Talk to your pharmacist. Ask if the generic you’re using has been tested in real patients. Check if your prescriber has a preference. Don’t assume approval = identical performance.
Regulators know the limits of pharmacokinetic studies. They’re updating guidelines. Scientists are building better tools. But until those tools become universal, the burden of proof still falls on the patient.
Are pharmacokinetic studies the only way to prove a generic drug works?
No. While pharmacokinetic studies are the most common method, regulators accept alternatives depending on the drug. For topical products, in vitro permeation testing (IVPT) or dermatopharmacokinetic methods are used. For some inhaled drugs, aerodynamic particle size analysis replaces blood tests. In rare cases, clinical endpoint trials - comparing actual patient outcomes - are required, especially for drugs with narrow therapeutic windows. The WHO even allows pharmacodynamic studies or in vitro tests for certain formulations.
Why do some generic drugs cause side effects when the brand didn’t?
Sometimes, it’s not the active ingredient - it’s the fillers. Excipients like dyes, preservatives, or coating agents can affect how the drug is absorbed or trigger immune reactions. Two generics can have identical pharmacokinetic profiles but different excipients, leading to different tolerability. This is especially true for patients with sensitivities, allergies, or gastrointestinal disorders. In rare cases, even minor differences in crystal structure or particle size can alter release rates - something blood tests might miss.
Can a generic drug pass bioequivalence testing but still be unsafe?
Yes. A 2010 PLOS ONE study showed that two generics of gentamicin passed all standard bioequivalence tests but had significantly different pharmacodynamic effects in patients - meaning one worked, the other didn’t. This happened despite identical chemical composition. The issue wasn’t absorption - it was how the drug interacted with bacterial targets. Pharmacokinetics measures what happens in your blood, not what happens at the site of action. For antibiotics, anticonvulsants, or anticoagulants, this gap can be dangerous.
How much does a bioequivalence study cost?
A full pharmacokinetic study typically costs between $300,000 and $1 million USD. The price depends on the number of subjects, study duration, complexity of the drug, and whether it’s tested under fasting and fed conditions. For complex formulations like extended-release tablets or inhalers, costs can rise even higher. The entire process - from formulation development to regulatory submission - usually takes 12 to 18 months.
Why don’t all countries use the same bioequivalence standards?
Regulatory systems vary based on resources, infrastructure, and risk tolerance. The FDA and EMA have detailed, science-driven guidelines. Many low-income countries lack the labs, trained staff, or funding to run complex studies, so they rely on simpler criteria - like dissolution testing or equivalence to a reference product from a trusted country. The WHO promotes harmonization through ICH M13A, but adoption is inconsistent. As a result, a generic approved in the U.S. may not be approved in Nigeria or Vietnam - even if it’s chemically identical.




9 Comments
Pharmacokinetic studies are a good start but they’re not the whole story. In India, we see generics that pass bioequivalence but still cause GI upset in patients-usually because of excipients. The FDA’s 80-125% range is a statistical band, not a biological guarantee. We need more focus on real-world outcomes, not just blood curves.
It’s funny how we treat drugs like math problems. You plug in Cmax and AUC, and out pops ‘equivalent.’ But the human body isn’t a lab. It’s a messy, adaptive, emotionally charged system. A pill that works for one person might fail for another-not because of chemistry, but because of sleep, stress, gut flora, or even the time of day. We’re reducing life to numbers because it’s easier than admitting we don’t fully understand it.
Oh honey. You think the FDA’s 80-125% rule is the gold standard? It’s a compromise between profit and public safety. The real gold standard is a 5-year randomized controlled trial with 10,000 patients. But who’s gonna pay for that? The generic industry? Please. They’d rather fund a 36-person fasting study and call it a day. And don’t get me started on the ‘BCS Class I’ loophole. That’s just regulatory cosplay.
So... you're saying that two pills that look identical, taste identical, and have the same active ingredient... might not be the same? Shocking. Next you'll tell me that two blue cars might have different engines. What's next? Maybe your left kidney works better than your right? This is why people don't trust science anymore.
I’ve been on generic levothyroxine for 8 years. My TSH was stable until my pharmacy switched brands. Suddenly, I was exhausted, gaining weight, and cold all the time. My doctor said ‘it’s bioequivalent’-but my body didn’t agree. I switched back to brand and felt human again. Don’t assume equivalence. Listen to your body. And if your pharmacist won’t let you choose? Ask for a prescription note. You’ve got rights.
Big Pharma and the FDA are in bed together. They want you to believe generics are safe. But what if the excipients are laced with microplastics? What if the coating is designed to trigger inflammation so you need more pills? I’ve seen the documents. They don’t test for long-term immune effects. They test for blood levels. That’s not medicine. That’s corporate math.
Just to clarify-when a drug is BCS Class I and the formulation is very similar, PBPK modeling is now accepted by the FDA as a substitute for human PK studies. It’s not theory anymore. It’s regulatory policy. For example, metformin and atorvastatin have had studies waived using this method. The system is evolving. It’s not perfect, but it’s moving beyond blood draws.
Y’all act like this is some new scandal. Nah. This is how capitalism works. If you can get away with a $300K study instead of a $10M clinical trial, you do it. The FDA knows. The WHO knows. The Indian manufacturers know. The only ones who don’t? The people swallowing the pills. So yeah-your blood test says ‘equivalent.’ But your body? It’s the last person who gets to vote.
USA and Europe think they’re so advanced with their fancy PK studies. But in India, we’ve been making generics for decades that save millions. We don’t need your 36-person trials. We have real-world data from 100 million patients. And guess what? Most of them are fine. Your fear-mongering about excipients? It’s just rich-country privilege. Your body doesn’t break because of a filler. It breaks because you’re stressed, eating junk, and sleeping 4 hours. Stop blaming the pill. Blame your lifestyle.